联系电话:
15902318117,13908347211
邮箱:lesch_nyhan_china@163.com
地址:重庆市九龙坡区谢家湾正街55号

Lesch-Nyhan 综合征(Lesch-Nyhan Syndrome, LNS),又称自毁容貌综合征,是由于次黄嘌呤鸟嘌 呤磷酸核糖基转移酶(Hypoxanthine guanine phos⁃ phoribosyl-transferase,HGPRT)失活或缺乏所导致 的先天性遗传代谢病[1] 。HGPRT酶功能为催化次 黄嘌呤转换为次黄嘌呤核苷酸(肌苷酸,IMP),也 可将鸟嘌呤转化为单磷酸鸟苷(GMP)[2] 。HGPRT 酶的失活或缺乏会导致次黄嘌呤、鸟嘌呤以及磷 酸核糖焦磷酸的聚集。过多的次黄嘌呤和鸟嘌呤 通过黄嘌呤氧化酶转化为尿酸,致使患者表现为 高尿酸血症或尿症[3] 。由于脑组织中不存在自5- 磷酸核糖至尿酸的从头合成嘌呤核苷酸的路径, 嘌呤核苷酸只能通过 HGPRT 作用的补救路径合 成,因此HGPRT酶缺乏影响神经系统的生长发育[4]。HGPRT 编码基因 HPRT1 位于 Xq26.2-q26.3,LNS 患者多为男性,由女性携带者传递,属 X染色体 隐性遗传病(OMIM 308000),发病率为 1/555 556 ~ 1/100 000[3,5-7] 。LNS具有广泛的临床表现,典型症 状包括高尿酸血症、精神发育迟缓、严重的运动 障碍、舞蹈手足徐动症 和 自我伤害行为,以及 HPRT1相关的痛风性关节炎、血尿[5,8-9] 。目前为止, 国内外学者在HPRT1基因已经发现超过400种致 病性突变,中国LNS患者已有报道,但此类患者婴 儿期特征的描述性研究甚少,或呈散发报道,临床 医生对婴儿期特点认识不足,从而延误诊断和治 疗。另外HPRT1基因型与婴儿期发病LNS表现型 的关系尚不清晰。因此,本文报道了4例婴儿期发 病LNS患者的临床与基因突变特点,结合国内外曾 报道的中国患者的相关资料,探讨婴儿期发病LNS 的临床特点,便于临床医生认识此类罕见病。
1 研究对象与方法
1.1 临床资料收集 收集 2017—2018 年在首都 儿科研究所附属儿童医院神经内科门诊确诊的来 自无关家系的疑似 LNS 患儿。患儿自出生 3个月 即表现运动发育迟缓伴随肌张力异常,血尿代谢 筛查、头颅磁共振成像(MRI)、脑电图均未见明显 异常,生化检查血尿酸均偏高。本研究通过首都 儿科研究所附属儿童医院医学伦理委员会批准 (伦审编号:SHERLL2013046)。患儿监护人均知 情同意,并签署知情同意书。
1.2 全外显子测序、变异筛选和家系分离验证等 抽取先证者及其父母静脉血各4 mL后提取基因组 DNA,对患儿进行全外显子测序(WES)。利用 公共数据库( 1k Genome[10] ,ExAC[11] ,gnomAD[12] 等)对基因变异进行变异人群携带率注释,同时使 用生物信息学软件(MutationTaster2[13] ,PROVE⁃ AN[14] ,Polyphen-2[15]等)对变异危害性进行注释, 确定基因变异后蛋白功能改变。对于隐性遗传的 基因,如果该基因变异最小等位基因频率(MAF) 在实验室内部数据库、东亚人群> 3%,则标注为 常见多态性变异;对于显性遗传的基因,如果该 基因变异 MAF> 1%,则标注为常见多态性变异;对于错义变异,如果有2个或2个以上软件预测该 基因变异具有危害性(deleterious effect), 则标注 为影响基因功能的危害性变异;将编码区±2位点 称为经典剪切位点,同时采用其他软件(Human Splicing Finder)寻找非经典剪切位点。根据以上 流程筛选出罕见危害性变异结合患者表型进行核 心家系成员(父母和同胞)验证,保留家系成员共 分离的变异位点。最终,根据《美国医学遗传学与 基因组学学会遗传变异分类标准与指南》评估变 异的致病性[16] 。通过制作 HGPRT 三维蛋白模型, 视觉化展示HPRT1基因致病性突变对于蛋白结构 的影响。
1.3 文献检索及筛选 在 Pubmed 中,以“Lesch- Nyhan Syndrome”、“HPRT1”为关键词进行文献检 索;在知网和万方数据库,以“LNS”、“ 自毁容貌 症”、“HPRT1基因”为关键词进行文献检索。阅读 文章摘要及全文筛选出截止 2020 年 12 月前报道 的婴儿期发病中国LNS患者的文献。
2 结果
2.1 4例婴儿期发病LNS患者临床特点 4例LNS 患儿均为男性,均在婴儿期发病,平均发病年龄 为 3.8个月,均以运动发育明显落后、肌张力异常 就诊,就诊年龄分别为 11、10、10月龄和2.5岁。生 化检测显示4例患儿血尿酸水平分别为482、464、
486、467 μmol/L,均高于均值237.0 ~ 356.9 μmol/L, 其中例 3 和例4伴肾结石、血尿。例4 在 1 岁左右 出现自我伤害行为,表现为反复咬口腔黏膜及手 指,咬伤处黏膜及皮肤出现溃疡。在后续随访中, 例2 和例 3分别在1岁后( 1岁余和14月龄)也出现 自我伤害行为。4例患儿在应用别嘌呤治疗后尿 酸均有所降低;左旋多巴与苄丝肼(美多芭)治疗
2个月肌张力改善不明显停用。例4(5岁)应用 S-腺苷甲硫氨酸(SAMe)治疗 3个月,肌张力障碍及 自残症状改善均不明显,停用。截至 2020年,4例 患儿中,仅例 1(2岁)未出现自我伤害行为或舞蹈 手足徐动症。
2.2 HPRT1基因突变特点 筛选的罕见危害性突 变位点仅剩HPRT1基因变异,家系验证显示患儿 母亲均为突变携带者(图 1),符合X染色体连锁隐 性遗传致病模式,其中2 个突变既往文献已有报 道 。遗传学家根据 ACMG 完成致病性进行评估 ——4个HPRT1变异都为致病性/疑似致病性变异 (NM_000194,NP_000185)(表 1),由于评估为致病 性变异,后面将改写为突变。图 2 展示了 HGPRT 蛋白突变前后的三维模型图,显示以上突变导致 该蛋白结构、酸碱性或极性发生改变。
2.3 文献检索结果 共筛选出9篇与HPRT1基因 相关 的 LNS 文献 ,包含 11 例婴儿期发病 的案 例[9,17-24] 。与 LNS 相关 的 HPRT1 基 因 突 变信 息 及患者临床资料详见表2。通过文献回顾发现,国 内 HPRT1 基因相关的婴儿期发病的 LNS 患者全 部为男性,发病年龄为 1.5 ~ 12.0 月龄,中位数
4 月龄。患者主要存在智力迟缓(100%,14/14)、 运 动 发 育 迟 缓(100%,15/15)、 高 尿 酸 血 症 (100%,15/15)、 自我伤害行为(93%,14/15)、 肌 张力障碍(85%,11/13)、 不自主运动或者舞蹈手足 徐动症(60%,9/15)、 血尿或结晶尿(47%,7/15), 部分患者具有异常脑 MRI、痉挛、语言发育落后、 肢体轻瘫等表型。
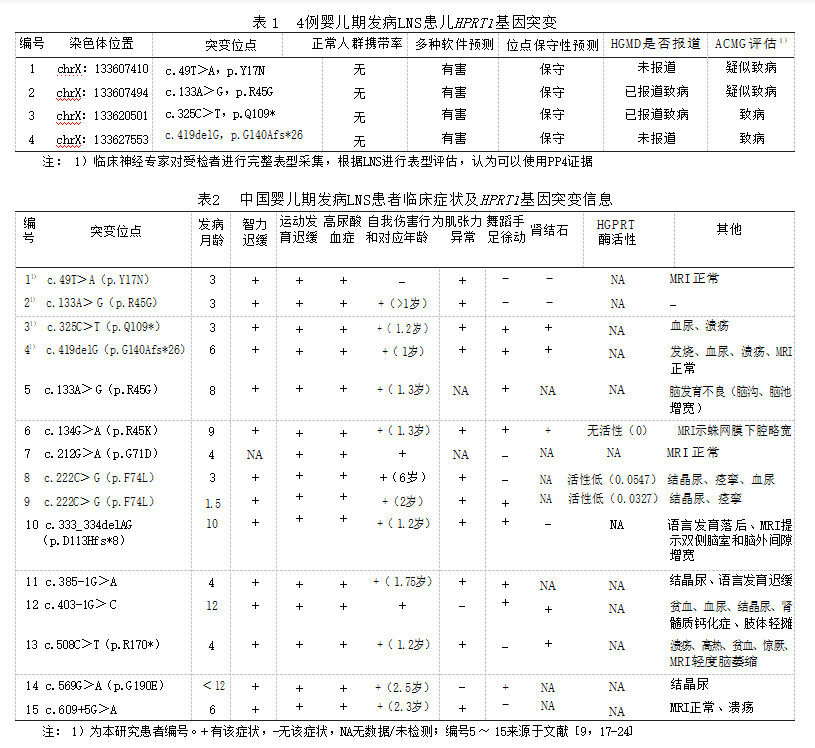
a 测序图和家系图;
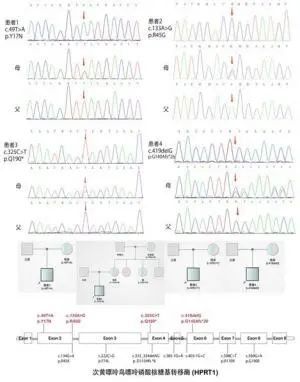
b、c 国内已报道的婴儿期发病的 LNS 患者携带的HPRT1突变位点图谱
图 1 4例婴儿期发病LNS患儿一代测序结果
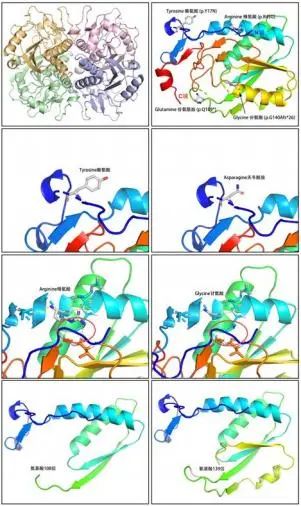
a HGPRT 酶四聚体,由四个完全相同的 HGPRT 单体构成;
b HGPRT单体存在 β-α- β 结构,蛋白 N端、C端与突变位点已标出(对照);
c、d HGPRT第 17位氨基酸,由酪氨酸变为天冬氨酸(p.Y17N);
e、f HGPRT 第 45 位氨基酸,由精氨酸变为甘氨酸(p.R45G), 精氨酸与 179 和 180位的缬氨酸和甘氨酸之间的极性相互作用消失,可能影响蛋白结构稳定性;
g 突变 p.Q109*导致蛋白在 109 位氨基酸处截短(与b 比较,结构不完整);
h突变p.G140Afs*26导致氨基酸从 140位开始序列完全改变(与b 比较,结构不完整)
图2 HGPRT 蛋白模型图
3 讨论
LNS是一种非常罕见的 X 连锁隐性遗传的神 经性疾病。患者所缺乏的嘌呤补救合成酶HGPRT 可催化次黄嘌呤转换为次黄嘌呤核苷酸,或将鸟 嘌呤转化为 GMP,在嘌呤核酸的生成中起着重要 的作用[3] 。HGPRT是由四个相同的亚基组成的四 聚体,每个单体的 β-α- β 结构上存在酶的活性位 点。基因突变可能破坏单体的二级结构影响活性 位点与酶作用物的结合,或者影响四聚体的结合, 从而造成酶失活[19] 。无效突变如无义突变和移码 突变,可造成蛋白的截短,影响蛋白的表达。HG⁃ PRT酶的失活或缺乏会导致次黄嘌呤、鸟嘌呤以及 磷酸核糖焦磷酸的聚集,导致尿酸升高,致使患者 表现为高尿酸血症或尿症[3] 。在脑组织中,嘌呤核 苷酸只能通过 HGPRT 所在补救路径合成,因此 HGPRT 缺乏也会影响神经系统的生长发育,导致 患者精神发育迟缓[4] 。但 HGPRT 的失活或缺乏是 通过何种分子机制导致神经功能障碍,仍待进一 步的探究[7]。
智力迟缓、运动发育迟缓、高尿酸血症是婴儿 期发病 LNS的中国患者的主要临床特点。其中高尿酸血症( 100%)和运动发育迟缓(100%)在国内婴儿期发病的 LNS 患者中出现的比例与国外既往报道(100%和91%)相似。但国内患者存在智力迟缓( 100%)比例高于国外报道(67%)。由于国内例数目限制,此结论有局限性,需要后期收集更多病例进行观察、分析和验证。本文中 4例均为男性 LNS 患儿,各自携带 HPRT1 基因致病性突变。4例出现高尿酸血症、精神发育迟缓和肌张力障碍,与国内外所报道 LNS 患者临床表型相符。其中,例 3 和例4症状较重,已出现出自我伤害行为,出现时间分别为 14 和 12月龄,并伴有肾结石、血尿、溃疡等症状;然而例 1 和例2,仅表现为高尿酸血症和智力低下,且都尚未出现舞蹈手足徐动症,二者整体症状相对较轻;后续随访时患者1在1岁多出现自我伤害的迹象。与此相对应,例 3、4 携带HPRT1 基因突变为无义突变(p.Q109*)和移 码突变(p.G140Afs*26), 导致蛋白质截短,完全破 坏了 HGPRT酶原有的空间构型和结构完整性,使 其完全失活;而例 1、2 携带错义突变(p.Y17N 和 p.R45G)仅为错义突变,对酶活性影响可能相对较 小,所以症状相对较轻。例 2 比例 1病情重,可能 因为突变 R45 坐落于磷酸核糖转移酶功能域,对 酶活影响相对 Y17 位置较大。由此,我们认为突 变的类型与 LNS 的临床症状的种类和严重程度可 能相关。
笔者整理分析 11例婴儿期发病的LNS案例报 道(表2), 同样发现携带截短型突变(无义或者经 典剪接位突变)者(例3、4、10、11、12、13)临床表 型涉及较为广泛,全部出现自我伤害行为并且行 为出现较早;而携带错义突变的 LNS 患者中,大 多数患儿症状相对较轻,临床表型涉及更为狭 窄,患儿无自我伤害行为或行为出现较晚较轻。其中R45(例2、5、6)和 F74(例 8、9)为突变热点, 分别在 3 例和 2 例中国患儿中出现。另外还发现 即使携带相同的基因突变,临床症状也存在个体 差异 。如例 2 与例 5 的突变完全相同(p.R45G), 例 6 和例 2、5 的突变位点相同、氨基酸变化不同 (p.R45G 和 p.R45K),但例 2 没有舞蹈手足徐动症 和肾结石,脑核磁也未显示异常,以上表型差异可 能和例2 年龄太小有关。但例 8 与 9 为同卵双胞 胎,都携带F74L 突变,例 9 的发病年龄、自我伤害 年龄仍比例 8 早。因此,笔者认为HPRT1 基因突 变位点和类型与婴儿期发病LNS 的临床症状相 关,但可能存在其他影响因素。考虑该疾病的罕 见性,仍需更多的案例来进一步探究HPRT1基因 突变与临床表型的联系。
对于 LNS 患者,以对症治疗为主。比如,通过 碎石术治疗肾结石;通过别嘌呤醇控制尿酸形成, 维持尿酸含量在正常水平,缓解高尿酸血症以及 痛风。本研究中,4例患儿均应用别嘌呤治疗,尿 酸有所降低,但左旋多巴治疗2个月肌张力改善不 明显停用。然而,对于 LNS相关的神经系统症状, 如智力运动发育落后、自我伤害行为等,目前并 没有统一的疗法。左旋多巴、卡比多巴等精神药 物曾被用于 LNS 患者的治疗,然而其效果目前仍 有争议[8] 。深部脑刺激(deep brain stimulation)也被认为是一种很有前途的缓解患者自我伤害和肌张力障碍的疗法[7] 。近年来,氨己烯酸(Vigabatrin)与 SAMe 曾被用于缓解 LNS 患者的神经症状。虽为个例,案例9 中的患者经过氨己烯酸治疗后,再无自我伤害行为[9] 。同样,SAMe合并别嘌醇(Al⁃lopurinol)曾使 LNS 患者的自我伤害行为显著降低,其中部分患者肌张力障碍也有明显缓解[25-29]。虽然在意大利患者的研究中,SAMe仅能缓解一部分患者的自我伤害行为(2/14,14.3%),并且加重了部分患者的症状(5/14,35.7%),但其仍为LNS有潜力且有研究价值的治疗方法[29] 。虽然在本研究中,一例患儿应用SAMe治疗3月,肌张力障碍及自残症状改善均不明显,停用。但值得注意的是,目前已报到的用药后自我伤害行为显著降低的患者,多数为亚裔7/11(63.6%,马来西亚、日本)。显示 SAMe对自我伤害行为无疗效或者会加重症状的案例全部来 自意 大利 的研究[30] 。这 暗示 了SAMe疗法或许在亚洲人种中药效更强,不过需要后续扩大患者数量进行严格的临床试验来证明其疗效。综上所述,本研究在4例婴儿期发病的LNS患者中发现了 4 种不同的HPRT1基因突变位点,完善了 LNS 患者的基因突变图谱。通过总结分析婴儿期发病共 15例 LNS 患者表型与突变,明确了婴
儿期发病的 LNS患者在的主要特点高尿酸血症合并智力落后、运动发育迟缓,截短型突变患者的症状和自我伤害行为出现较早。本研究说明,对于此类临床表现患者,需尽早进行 LNS 致病基因测序(重点关注HPRT1基因),将有助于早期 LNS诊断治疗。
参考文献
[1] Huang J,Zhang C,Guo Q,et al. Lesch-Nyhan syndrome in a Chinese family with mutation in the hypoxanthine-guanine phos⁃phoribosyltransferase gene[J]. Clin Lab,2018 ,64(1): 197-200.
[2] Ng N,Kaur A,Shenoy M. Recurrent kidney stones in a child with Lesch-Nyhan syndrome:Answers[J] . Pediatr Nephrol,2019,34(3):425-427.
[3] Torres RJ,Puig JG. Hypoxanthine-guanine phosophoribosyl⁃transferase(HPRT) deficiency:Lesch-Nyhan syndrome[J]. Or⁃phanet J Rare Dis,2007,2:48.
[4] 黄佼 . Lesch-nyhan综合征 1例报道——来自HGPRT基因突变的中国家庭[J]. 中国生化药物杂志,2017,37(6): 435-436
[5] Arhakis A,Topouzelis N,Kotsiomiti E,et al. Effective treatment of self-injurious oral trauma in Lesch-Nyhan syndrome:A case report[J]. Dent Traumatol,2010,26(6):496-500.
[6] McCarthy GT,Green EM,Ogunbona O,et al. A population study of Lesch-Nyhan disease in the UK[J] . Dev Med Child Neurol, 2011,53( 1):34-39.
[7] Bell S,Kolobova I,Crapper L,et al. Lesch-Nyhan syndrome:Models,theories,and therapies[J]. Mol Syndromol,2016,7(6):302-311.
[8] Visser JE,Schretlen DJ,Bloem BR,et al. Levodopa is not a use⁃ful treatment for Lesch-Nyhan disease[J]. Mov Disord,2011,26 (4):746-749.
[9] Mak BS,Chi CS,Tsai CR,et al. New mutations of the HPRT gene in Lesch-Nyhan syndrome[J]. Pediatr Neurol,2000,23 (4):332-335.
[10] 1000 Genomes Project Consortium,Auton A,Brooks LD,et al.A global reference for human genetic variation[J]. Nature,2015, 526(7571):68-74.
[11] Lek M,Karczewski KJ,Minikel EV,et al. Analysis of protein-coding genetic variation in 60,706 humans[J]. Nature,2016, 536(7616):285-291.
[12] Karczewski KJ,Francioli LC,Tiao G,et al. The mutational con⁃straint spectrum quantified from variation in 141,456 hu⁃ mans[J] . Nature,2020,581(7809):434-443.
[13] Schwarz JM,Cooper DN,Schuelke M,et al. MutationTaster2:mutation prediction for the deep-sequencing age[J]. Nat Meth⁃ ods,2014,11(4):361-362.
[14] Choi Y,Chan AP. PROVEAN web server:A tool to predict the functional effect of amino acid substitutions and indels[J]. Bio⁃ informatics,2015,31(16):2745-2747.
[15] Adzhubei I,Jordan DM,Sunyaev SR. Predicting functional ef⁃fect of human missense mutations using PolyPhen-2[J]. Curr Protoc Hum Genet,2013,76( 1):1-41.
[16] Richards S,Aziz N,Bale S,et al. Standards and guidelines for the interpretation of sequence variants:a joint consensus rec⁃ ommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med,2015,17(5):405-424.
[17] Hou JW. Atlantoaxial subluxation with recurrent consciousness disturbance in a boy with Lesch-Nyha
[19] Liu N,Zhuo ZH,Wang HL,et al. Prenatal diagnosis based on HPRT1 gene mutation in a Lesch-Nyhan family[J]. J Obstet Gynaecol,2015,35(5):490-493.
[20] Yang MT,Mak SC,Chi CS,et al. Lesch-Nyhan Syndrome:re⁃ port on two brothers[J]. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi,1994,35(6):552-558.
[21] 田小娟,丁昌红,代丽芳,等 . HPRT1基因突变致 Lesch-Ny⁃ han综合征 1 例临床特点及基因分析[J]. 中华实用儿科临 床杂志,2019,34(18):1424-1426.
[22] 赵勇,金炳旭,钱旭光 . 婴幼儿Lesch-Nyhan综合征3例的临 床特征及HPRT1基因分析[J]. 中国儿童保健杂志,2020, 28(7):834-836.
[23] 李小丽,贾天明,张晓莉,等 . 2例 Lesch-Nyhan综合征患儿及家系临床表型和基因突变分析[J]. 中华实用诊断与治疗 杂志,2020,34(2):157-159.
[24] 辛庆刚,赵澎,舒剑波,等 . 早期诊断 Lesch-Nyhan综合征 1 例[J]. 中国实用儿科杂志,2020,35(10):822-824.
[25] Glick N. Dramatic reduction in self-injury in Lesch-Nyhan disease following S-adenosylmethionine administration[J]. J Inherit Metab Dis,2006,29(5):687.
[26] Momosaki K,Kido J,Matsumoto S,et al. The effect of S-adeno⁃ sylmethionine treatment on neurobehavioral phenotypes in Lesch-Nyhan disease:A case report[J]. Case Rep Neurol, 2019,11(3):256-264.
[27] Togawa M,Saito Y,Maegaki Y,et al. Treatment of self-injuri⁃ ous behaviors in Lesch-Nyhan syndrome with S-adenosylme⁃ thionine[J]. No To Hattatsu,2017,49( 1):25-27.
[28] Chen BC,Balasubramaniam S,McGown IN,et al. Treatment of Lesch-Nyhan disease with S-adenosylmethionine:experience with five young Malaysians,including a girl[J]. Brain Dev, 2014,36(7):593-600.
[29] Lauber M,Plecko B,Pfiffner M,et al. The effect of S-adeno⁃ sylmethionine on self-mutilation in a patient with Lesch-Ny⁃ han disease[J]. JIMD Rep,2017,32:51-57.
[30] Dolcetta D,Parmigiani P,Salmaso L,et al. Quantitative evalua⁃ tion of the clinical effects of S-adenosylmethionine on mood and behavior in Lesch-Nyhan patients[J]. Nucleosides Nucle⁃ otides Nucleic Acids,2013,32(4):174-188.
